Présentation et diagnostic des angio-oedème héréditaires (ou oedèmes angioneurotiques héréditaires)
Ils sont à distinguer des angio-oedèmes « allergiques » de mécanisme anaphylactique ou anaphylactoïde qui s'accompagnent généralement de manifestations urticariennes.
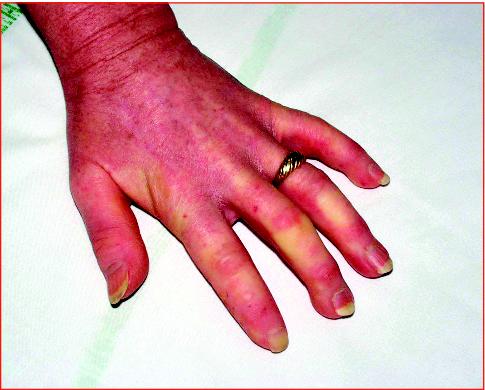
Les oedèmes angioneurotiques héréditaires se manifestent par des épisodes récidivants, sans éléments urticariens. Les premières manifestations se font généralement dans l'enfance avec majoration des signes à la puberté. La présentation peut se faire par des épisodes de gonflement de la face mais aussi des mains, des bras, des jambes, des pieds, de la langue, du larynx, des viscères, expliquant alors des épisodes de douleurs abdominales récurrentes faisant parfois errer le diagnostic. Il s'écoule en règle plus de 10 ans entre les premières manifestations et l'établissement du diagnostic. Les crises peuvent être déclenchées par des circonstances de stress (traumatismes, interventions chirurgicales, soins dentaires). Le caractère familial aide au diagnostic. Il s'agit cependant d'une mutation de novo dans un quart des cas. Il s'agit d'une maladie autosomique dominante.
On distingue deux grands types d'angio-oedème héréditaires :
- le type I (environ 85%) où il existe un déficit quantitatif en inhibiteur de la C1 estérase (C1 INH)
- le type II (environ 15 %) où le dosage est normal mais le C1 INH présente des anomalies fonctionnelles
On décrit un type III plus marginal sans anomalie quantitative ni fonctionnelle du C1 INH où le taux d'oestrogènes semble impliqué.
Le dosage quantitatif du C1 INH est abaissé dans le type I. Si ce dosage est normal alors que le diagnostic est suspecté, on peut alors procéder à un dosage « fonctionnel ». Si le dosage du C1 INH est nécessaire pour l'établissement du diagnostic chez des patients ciblés, le dosage de la fraction C4 du complément est utile en terme de dépistage, car le taux de ce marqueur est en règle abaissé en permanence ou au décours d'une crise.
Principes de prise en charge
Au cours de la crise, la gravité est liée au risque asphyxique qui pourrait expliquer 30 % des décès de ces patients. L'existence de signes de détresse respiratoire impose le recours au contrôle des voies aériennes par intubation voire trachéotomie.
L'utilisation des antihistaminiques et des corticoïdes est sans efficacité dans cette pathologie.
L'efficacité de l'adrénaline n'est pas prouvée (efficacité non constante et transitoire qui permettrait éventuellement d'éviter l'intubation dans certains cas).
Le traitement des formes graves repose sur l'utilisation de concentré de C1 INH (500 à 2000 unités) qui permet une résolution des signes dans l'heure.
Des traitements validés sont proposés pour limiter la récurrence des crises (traitements à base d'androgènes ou de molécules anti-fibrinolytiques). L'injection de concentré de C1 INH peut être envisagée en prophylaxie préopératoire.
L'utilisation de traitements comme les IEC et les contraceptifs à base d'oestrogènes sont à éviter.
Les patients sont invités à porter sur eux une carte spécifiant qu'ils sont atteints de cette pathologie.


 Rappel:
Rappel: