En France, cette famille regroupe : l'anis vert, la berce, la carotte, le cerfeuil, le céleri, le persil, le panais, le fenouil, la coriandre, le cumin, l'angélique, mais aussi : la ciguë, le criste marine, les panicauts, les œnanthes, les buplèvres, les aches et les lasers.


 Rappel:
Rappel:
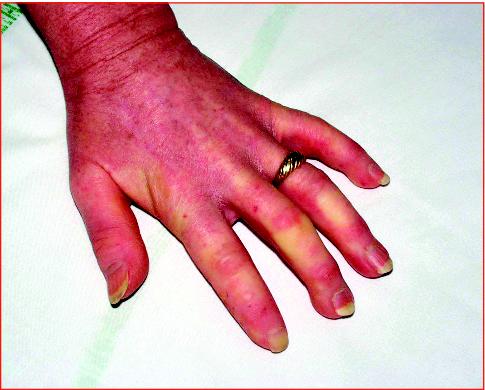

Systemic lupus erythematosus (SLE) is a chronic, inflammatory autoimmune disorder. It may affect the skin, joints, kidneys, and other organs.
Symptoms vary from person to person, and may come and go. The condition may affect one organ or body system at first. Others may become involved later. Almost all people with SLE have joint pain and most develop arthritis. Frequently affected joints are the fingers, hands, wrists, and knees.
Inflammation of various parts of the heart may occur as pericarditis, endocarditis, or myocarditis. Chest pain and arrhythmias may result from these conditions.
General symptoms include:
- Arthritis
- Fatigue
- Fever
- General discomfort(malaise)
- Joint pain and swelling
- Muscle pain
- Nausea and vomiting
- Pleural effusions
- Pleurisy (causes chest pain)
- Psychosis
- Sensitivity to sunlight
- Skin rash -- a "butterfly" rash over the cheeks and bridge of the nose affects about half of those with SLE. The rash gets worse when in sunlight. The rash may also be widespread.
- Cold sensitivity
Additional symptoms that may be associated with this disease:
- Abdominal pain
- Blood disorders, including blood clots
- Blood in the urine
- Coughing up blood
- Hair loss
- Mouth sores
- Nosebleed
- Numbness and tingling
- Red spots on skin
- Skin color is patchy
- Swallowing difficulty
- Visual disturbance







 Hématologique (pancytopenie)
Hématologique (pancytopenie)



